Proteins fold into shapes that function in the cell
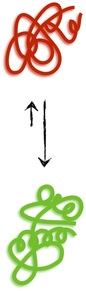
Proteins are polymers. More specifically, they are chains of amino acids, of which there are twenty different types denoted with letters like V or A or Q. Thus, a typical protein has an amino acid sequence that runs on for hundreds of letters like VLSMEAG . . . The different properties of the amino acids (which span the gamut: charge, size, bendiness etc.) lead proteins with different sequences to interact differently with their environment. The result is that, in general, a protein in the cell will fold up into a quite specific, functional shape called its native conformation (green, bottom) that is favored energetically over the vast, disorderly ensemble of other shapes (red, top) that cannot accomplish the biological purpose(s) of the natively folded protein.
Hydrophobicity drives the folding of many proteins
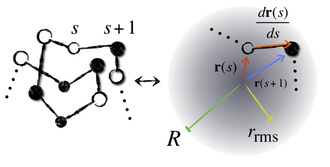
Just like oil separates from water, some amino acids try to hide themselves from the watery surroundings of the cellular medium by burying themselves inside the core of the "globule," that is, the crumpled up ball of yarn that the protein forms when in solution. The result is a competition among different parts of the protein chain for the space in the globule's core. We work with a simple model of this competition that just keeps track of how far away each amino acid is from the center of the globule and from its neighbors on the chain, while the chain is constrained to stay reasonably well-spread out over the globule as a whole. This allows us to get shape data from sequence by computing burial traces like the ones below:
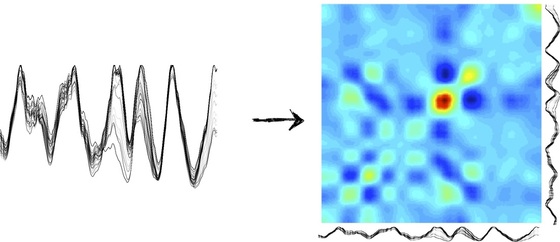
Burial fluctuations allow us to explain allostery
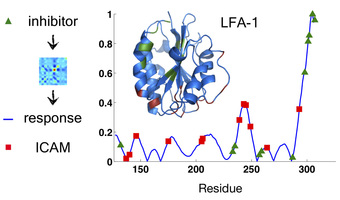
On the left-hand side of the above picture, we have many different low-energy "burial traces" for the protein LFA-1 near its native shape. This is telling us from our model, moving from left to right, that the beginning of the chain wants to be in the globular core, and then 20 or so amino acids in you get un-buried, and then further along you get buried again, and so on. Notice, though, that there's a lot of variability in the burial in certain parts of the protein. Moreover, as burial across the whole protein fluctuates, certain parts of the chain tend to move in tandem, and we can make a correlation color map of this motion (right hand side). As a result, we know which motions in one part of the protein are likely to produce motions in which other parts. We can use this information to learn many things about a protein, such as what sorts of "allosteric motions" it will tend to undergo. In the adjacent figure, the burial covariances of the protein LFA-1 were used to predict what motions the chain would undergo upon binding of an inhibitor drug to a known binding site (green). The resulting blue trace matches well with the ICAM protein-protein interface (red) that is known to be disrupted by drug binding.
There's much more to do!
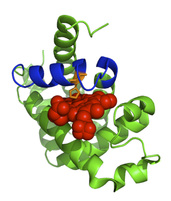
Allostery is just one of many phenomena that may be understood better by using burial mode analysis to describe the physics of conformational fluctuations in real proteins. To the left, the sequence of sperm whale myoglobin was used to compute from burial modes the region of highest structural variability in the protein. It turns out that this region (colored blue) is the helix that contains His 93 (orange), the amino acid needed to chelate the protein's co-factor, heme (red). Whether in cases of ligand binding (like this one), or in ones of phosphorylation, mutation, misfolding, or aggregation, we are interested in applying the burial mode model to real proteins whose properties have real implications for drug-design, neurodegenerative disease, and cancer.
